Javascript message
This site uses javascript. If javascript is disabled in your browser, some functionality may be impaired.
Frequently Asked Questions
Yes, the columns can be operated under normal phase, polar mode and reversed phase conditions. The regular columns are packed to give highest possible column efficiency under normal phase and polar mode conditions (alcohol or acetonitrile, no alkane). They can be converted to reversed phase via 100% ethanol or isopropanol as an intermediate solvent. Of course, the columns can also be re-conditioned to the original mode. By converting the column, one will observe a loss in column efficiency when comparing the plate count in RP vs. NP mode. For regular use under RP conditions, we recommend the purchase of columns that are packed giving highest possible efficiency under reversed phase conditions, AmyCoat RP and CelluCoat RP.
The reversed phase Kromasil columns C4, C8 and C18 should be operated with at least 5% organic modifier. Under 100% aqueous conditions, the columns are likely to undergo dewetting which means the mobile phase will be expelled from the porous system due to surface tension. In this case the analytes elute more or less with the void volume. The problem arises mainly if the flow is stopped. If a column has been dewetted, it can easily be restored by pumping mobile phase with at least 50% organic modifier through the column for ca 40 min.
Kromasil C18(w) is specialised phase variant that is fully compatible with 100% aqueous conditions. Find out more about it on the product page.
Column plugging is responsible for well over half of all column failures. Therefore thorough filtration (0.2 μm) of mobile phases and samples is a key for long column lifetime.
The mobile phase compatibility is another important factor. Standard RP stationary phases are stable under pH 1.5 to 9. Under certain conditions (organic buffers) the stationary phases are even chemically stable up to pH 12. Please consult with our technical support if you would like to use your classic Kromasil column at pH values exceeding pH 9. Generally, organic buffers are milder towards the silica based stationary phase than inorganic ones, such as phosphates or carbonates. If you need to run your method at high pH choose Kromasil EternityXT which is stable up to pH 12.
Further, the columns should never be left in a buffered mobile phase when the flow is interrupted for more than 10 minutes. Either choose a very low flow rate (0.1 ml/min for a 4.6 mm ID column) instead of stopping the flow completely, or exchange any buffered mobile phase with non-buffered one (e.g. water/acetonitrile 70/30). This will prevent the column from being clogged with precipitated buffer salts.
The performance is best checked by re-running the separation shown on the enclosed test chromatogram. The exact conditions are mentioned. The sample amount should not exceed the following quantities:
| Column Ø
[mm ID] |
test sample
[µg] |
| 2.1 | 2 |
| 4.6 | 10 |
| 10 | 50 |
| 21.2 | 225 |
In order to achieve representative plate counts, it is important to reduce the extra column volumes, e.g. correct tubing has to be installed between injection and the column and from the column to the detector.
An increased back-pressure, altered retention times and loss of column performance are all symptoms of deposits in the column or on the surface of the stationary phase. Most of the times, these problems can be overcome by the use of a correctly applied washing procedure. What should also be kept in mind is that in most cases the sooner washing (regeneration) of the column is performed, the better.
Strongly adsorbed species are collected at the solvent-inlet end of the column and in many cases it is a benefit to use a reversed flow during washing. Worth mentioning is that a well packed column should not loose performance as a result of reversed flow.
RP (Hydrophobic Phases)
Deposits are most commonly present as surface adsorbed species or precipitations.
| Suspected Deposition/Impurity | Recommended solvents | Examples |
| Lipohilic | Strongly Lipophilic solvents | Al, Tol |
| Polar (Small peptides) | Versatile solvents | DCM, THF, DMF |
| Strongly Polar / Ionic (water sol.) | Aqueous solvents | 50:50 DMF/w, THF/w |
| Polar, positively
charged (amines) |
Ion-exchange supressing mixtures | DMF/AA (1%), DMF/TFA (0.1%) |
| Macromolecular Depositions (Protein/ Large Peptide precipitations) | Strongly interaction breaking mixtures | DMF/1% SDS(aq), ACN/1% SDS(aq), Alc/AA, Alc/NEt3 (0.1%) Alc/10-100mM aqueous NaOH* |
| Salt/Buffer
precipitations |
Highly aqueous mixtures | Alc (10%)/w |
|
Al = Alkanes,
Tol = Toluene, DCM = Dichloromethane, THF = Tetrahydrofuran, DMF = Dimethylformamide, AA = Acetic acid, TFA = Trifluoroacetic acid, ACN = Acetonitrile, Alc = Alcohol (MeOH, EtOH), w = water, NEt3 = Triethylamine, SDS = Sodiumdecylsulphate * Use as last measure for not more than 10 column volumes, also end by acidifying the phase sing Alc/1% aqueous AA 50:50. |
||
Regeneration protocol
- Choose solvent/solvent mixture based on impurity. If not known, assume strongly polar / ionic species in order to avoid further precipitation.
- Use a low flow rate (10% of normal flow rate), possibly in reversed flow mode and at slightly elevated temperatures (< 40°C), for up to ten column volumes.
- If problems with back-pressure, occasionally check progress at normal flow.
- If the problem remains apply conditions for polar, positively charged and/or macromolecular depositions. If SDS is used, wash thoroughly with pure THF, DMF or ACN afterwards.
- If problems persist please consult our technical support for further advice.
NP (Polar Phases)
For polar phases, especially bare silica, very strong adsorption of polar residues is common. While under normal phase conditions highly polar impurities are not eluted, however the solution to this is the use of high levels of protic solvents, possibly in combination with acid.
If there are problems with altered retention times, first check the phase - water (moisture) equilibrium. The use of dry solvents could gradually "dry" the stationary phase, resulting in altered retention times. This is especially rue for bare silica.
Regeneration protocol?
- Eqilibrate the column using an aqueous-lipophilic mediating solvent such as THF.
- Use a low flow rate (10% normal flow rate) of pure Alcohol (MeOH), possibly in combination with acid (Acetic acid or Formic acid, 1-5%), for up to ten column volumes.
- If the problem persists, use a mixture of Alcohol and water, with added acid, under the same conditions as in 2.
- As a final measure: 0.5-1.5% NH3(aq), followed by 0.5-1.5% aqueous acid (HCl, AA etc.), water, alcohol, THF and finally the actual eluent.
- If problems persist please consult our technical support for further advice.
?: While using highly polar solvents, especially for bare silica, the stabilization of the system after washing could take a significant amount of time. This is due to the silica surface - water equilibrium being distorted.
Buffered mobile phases can basically be prepared in two ways:
- preparing the buffer, including pH adjustment prior to mixing with organic modifier.
- mixing buffer and organic modifier prior to pH adjustment.
While the pH is a well defined parameter in aqueous systems, it is not as straight forward to define or measure proton concentration in partially organic solvent systems.
It is therefore recommended to prepare the aqueous buffer, pH adjust it, filtering it through 0.4 µm and mixing it thereafter with the organic modifier. Attention should be given to the following points:
- buffers are effective within +/- 1 pH unit from their pKA ⇒ choose correct buffer system
- consider the risk of precipitation if high buffer concentrations are mixed with organic solvents
- always filter buffers through 0.4 mm filter prior to use
- be aware of the risk for bacterial or fungal growth in pure aqueous buffers
- the pH generally shifts upon the addition of an organic modifier (pH increases for inorganic
buffers (e.g. phosphates) and decreases for organic buffers (e.g. acetates)
Example: phosphate buffer pH 7.6 ⇒ addition of 40% MeOH ⇒ pH 8.5
Correct column storage is a prerequisite for problem free chromatography and a long column life time. Normal phase columns should be stored in heptane/2-propanol or another inert solvent that is free from additives.
Reversed phase stationary phases should be kept in a mixture of water / organic modifier, free from any buffers or additives that might precipitate during storage. Thus it is recommended to wash the column whenever chromatography is going to be stopped for more than 15 min with water / organic modifier 50/50, independently on whether the column will be detached from the HPLC instrument or not. If chromatography is only to be interrupted for a couple of hours it might be less laborious to keep the buffer containing mobile phase running at a very low flow rate. If the column is detached, close the inlet and outlet with a screw in order to avoid the stationary phase to dry out.
All HPLC columns should be stored at ambient temperature (18-26°C), without risk for mechanical shock, preferably in the original column box.
With each Kromasil column, in the column box, an individual test chromatogram is provided that shows the efficiency under a standard test conditions. If lost, a copy of this test chromatogram can be requested using a dedicated request form.
Keywords: Test Chromatogram, Certificate of Analysis, CoA
1. Mobile phase composition
Most reproducible results are obtained by weighing the mobile phase mixtures. For water / ethanol 30/70 (v/v) 300.0 g (300 ml x 1.0 g/ml) water are mixed with 546.0 g (700 ml x 0.78 g/ml) ethanol.
Especially when working with alcohols as polar modifier, online mixing by the HPLC pumps should be avoided, as the viscosity of the components varies significantly and hinders proper blending.
Mobile phases should always be prepared fresh, as evaporation will alter the composition of a two component mobile phase.
2. Shifting temperature
A difference of 5°C can render +/- 10% shift in retention times. It is therefore recommended to work with a column oven even when working under ambient temperature.
3. pH Control
The retention time of acidic and basic analytes depends upon the pH value. 0.1 pH unit can render a shift in retention time by 10%. It is therefore recommended to work at a pH value at least 2 pH units from the pKA.
4. Incomplete equilibration
It is recommended to equilibrate reversed phase columns with at least 10 column volumes, for buffered mobile phases with at least 20 column volumes before an analysis is initiated. Make sure all mobile phase channels are purged with the adequate mobile phase. Normal phase columns can require up to 10x longer equilibration times than reversed phase columns. Bare silica columns might take hours to equilibrate, special attention should be given to the water content in the normal phase systems. Most reproducible results are often obtained when deliberately adding 0.05% water the mobile phase.
5. Dewetting
Highly polar mobile phases can be expelled from the hydrophobic porous system, leading to less and less accessible surface area, and therewith causing shorter and shorter retention times. See also: "Can the column be operated with 100% aqueous mobile phase".
End capping describes the process of reacting silanol groups that have not been derivatized, e.g. octadecylchlorosilane. The end capping reagent is generally a smaller silane (e.g. trimethylchlorosilane) that is used to react with some of the residual hydroxyl groups. This treatment reduces the amount of available silanol groups that can undergo often undesired interactions with polar or charged analytes.
HILIC means Hydrophilic Interaction Liquid chromatography and is a technique used for separating highly polar substances. It can also be described as aqueous normal phase chromatography. The stationary phase has to be of polar character, e.g. silica, diol, amine, amide or zwitterionic. As a mobile phase, generally acetonitrile / water mixtures are used, where water is the strong component, thus exhibiting a higher elution strength than acetonitrile. Thus, HILIC is a sub-category of normal phase chromatography and shows opposite elution pattern to reversed phase.
The use of mobile phases with a low content of organic modifier (< 5 %) generally leads to what is known as "phase collapse". The latter is best described as a dewetting phenomenon, in which the highly aqueous mobile phase is excluded from the hydrophobic pore system due to surface tension. The effect is observed predominantly with reversed phase packing materials with high ligand density (> 3.2 μmol/m2). The loss of wetted surface results in a decrease in accessible interaction sites for the solute, and leads to a loss in retention and / or reduced loading capacity. The process can easily be reversed by purging the packed bed with a high content of organic modifier (> 50%) The phenomenon is most dominant for C18 and C8 packing materials with pores < 150 Å, but even with C4 modified packing materials, 100% aqueous mobile phases should be avoided in order to assure robust chromatography.
Kromasil C18(w) is specialised phase variant that is fully compatible with 100% aqueous conditions. Find out more about it on the product page.
For each particle size, there is an optimal linear flow rate with respect to column efficiency, according to the van Deemter plot.
| 3.5 µm | 5 µm | 10 µm | 13 µm | 16 µm | |
| Linear flow rate
(cm/min) |
7.1 | 4.7 | 3.3 | 2.4 | 1.9 |
| 2.1 mm ID | 0.3 | 0.2 | - | - | - |
| 4.6 mm ID | 1.5 | 1.0 | 0.7 | 0.5 | 0.4 |
| 10 mm ID | - | 4.7 | 3.7 | 2.4 | 1.9 |
| 21.2 mm ID | - | 21 | 15 | 10 | 8.5 |
For shorter separation times, the flow rate can however be increased until the pressure drop limit of the HPLC pump is reached, e.g. ca 400 bar.
The dead volume can also be called extra column volume and consists of the volume of a HPLC system between the point of injection to the point of detection, but excluding the column itself. Thus it includes the injection volume, the volume of the injector, the volume of the connecting tubing before and after the column, the volume of the end-fittings and frits, and the volume of the detector flow cell. The dead volume can be measured by replacing the column with a zero-volume connector. By injecting a very small sample amount, the time can be measured between the moment of injection and the maximum peak height. This time multiplied by the flow rate gives you a good estimate for the system dead volume.
The dwell volume or gradient dwell volume is responsible for the time delay of the gradient. It describes the volume of a gradient HPLC system between the mixing chamber and the column inlet. This volume does of course also exist under isocratic elution, but in that case, it has no impact on the separation. The gradient dwell volume comprises of the volume of the gradient mixer, the connecting tubing to the pump, the pump head and check-valves, the tubing between the pump and the injector, the injector itself, and the tubing between the injector and the column inlet. When initiating a gradient, the column is not subjected to the change in eluent composition until the gradient has passed the dwell volume. During that time, the column is operated under isocratic elution. Attention has to be given when transferring a gradient method from one instrument to another. If the dwell volume differs, the retention times will likely differ as well, despite an identical method and column. The dwell volume can be measured by running a step gradient from 100% methanol to 100% methanol + 10 mg/L acetophenon. The UV detector will detect an S-shaped detector trace. The dwell volume is equal to the time between the injection and half height of the detector trace, multiplied by the flow rate.
In both cases, the stationary phase is more hydrophobic than the mobile phase. The hydrophobicity of RP-stationary phases is generally higher than that of HIC media. Elution in RP is obtained by adjusting the polarity of the mobile phase with a water miscible solvent, e.g. an alcohol or acetonitrile. Elution takes place either under isocratic (constant modifier concentration) or a under gradient conditions (increase in modifier concentration).
HIC media has weaker hydrophobic character and elution is induced by decreasing the polarity in the mobile phase by virtue of a decrease in salt concentration. HIC is generally applied in protein separations, where exposure to organic solvents under prolonged time might induce irreversible unfolding, thus loss of biological activity of the proteins.
For analytical purposes, RP-HPLC is an unchallenged method when it comes to separating closely related substances, including proteins.
There is virtually none! Both the polysaccharide based CSPs Kromasil AmyCoat and CelluCoat, as well as the CSPs based on a crosslinked network polymer, Kromasil TBB and DMB, are based on highly mechanically stable, in-house produced, porous silica spheres. Moreover the proprietary coating method for the polysaccharide based phases makes these CSPs highly resistant to hydrodynamic forces. Thus, all Kromasil CSPs can be operated up to 400 bar back pressure. Exploiting the full pressure drop range of classical HPLC instruments allows for fast analysis and moreover very efficient column equilibration at high flow rates.
The shipping solvent is stated on the test chromatogram and depends upon the surface modification of the stationary phase.
When pumping mobile phase through a particular bed, a certain pressure drop δP over the column will result. The relationship between particle diameter dp and the flow velocity u is given by the Darcy´s law for non-compressible solvents:

Accordingly, δP is proportional to the linear velocity u, the column lengths L and the viscosity of the mobile phase η, and inversely proportional to the constant k0 and the square of the particle diameter dp. The constant k0 is a function of the interstitial porosity εi according to the Karman-Cozeny equation:
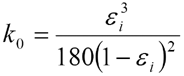
The equation above shows that the interstitial porosity εi has a tremendous influence on the permeability of the bed and therewith also on the pressure drop. For a well packed reversed phase be the interstitial porosity can be assumed to be 0.33 which leads to k0 = 4.45.10-4. The column permeability (dp2·k0) does in practice depend entirely upon the particle diameter and the particle size distribution. The latter is a very important parameter, since the particles of an HPLC packing material are never mono-disperse.
Broad distribution has two negative effects:
- the largest particles control the efficiency and
- the smallest particles control the pressure drop.
Hence, broad particle size distribution should be avoided.
Capillary tubing
When working with analytical HPLC instruments, capillaries with external diameter of 1/16" (1.6 mm) are generally used to connect the different parts of the instrument. Those parts that do not affect the extra column volume should have internal diameters (I.D.) of 1 mm. They bring along little risk for clogging and show minimum flow resistance, which contributes to the extra column pressure drop. For those connections critical for the extra column volume, internal diameter of 0.17 mm is recommended. Such capillaries add only 23 μL extra volume per meter of tubing (1.0 mm ID tubing: 800 μL/m). In order to avoid clogging of such fine capillaries, it is of uppermost importance to filter both, mobile phases and sample solutions.
For preparative columns the tubing has to be scaled up in order to reduce the flow resistance caused by the higher flow rates (δPcap≈dcap-4):
| Column I.D.
mm |
Tubing I.D.
mm |
|---|---|
| 20 | 0.25 |
| 50 | 0.5 |
| 100 | 1.0 |
Fittings
Fittings are connecting the various parts of the HPLC instrument. Steel fitting usually have a separate ferrule, some designs even use two ferrules, a ring and a conical piece. When tightened the first time, the ferrule is pinched permanently and the surrounded capillary is slightly compressed. The ferrule can no longer be removed from capillary. Plastic (PEEK) fittings are one single piece (nut, ferrule), and are installed by finger tightening only. Over tightening (common with steel fittings) must be avoided, since excessive force is likely to damage the threading and the capillary. It is recommended to tighten the screw only slightly with a wrench and to test if the connection is pressure resistance, meaning, no leakage occurs once mobile phase is pumped through the system.
The distance from end of tubing to ferrule lowermost end is depends upon the column and/or instrument design. Some examples are given below:
- 0.130" in case of Waters columns
- 0.80" for Valco design
- 0.170" for Rheodyne injector design
A guard column can be useful if the injection sample consists of a complex matrix with partially unknown content. However, a guard column does never make sample filtration redundant! If a guard column is to provide adequate protection for the analytical column, it must be changed often enough in order to avoid the column performance from deteriorating. By monitoring plate number (N), pressure, and resolution (Rs), the performance of the guard column, as well as the analytical column, can be assessed and a suitable moment for exchanging the guard column can be chosen.
As a rule of thumb, the guard column should be replaced when one of the following points applies:
- N decreases by > 10%
- pressure drop increases by > 10%
- Rs changes by > 10%
- more than 150 samples are injected.
Read more about ordering and handling guard columns on the guard columns page.
In reverse phase HPLC it is recommended to run a scouting gradient if the conditions for a successful separation are unknown. Such a run is performed from ca 10-80% organic modifier during 30-50 min, using a linear increase in elution strength. Based on the result of this scouting gradient, either an appropriate mobile phase composition can be chosen for isocratic elution (small molecules < 1000 g/mol) or a suitable gradient for the separation of peptides or other larger molecules.
Gradient elution can also be chosen for separation of compounds that vary greatly in their polarity. As a rule of thumb, if peaks can be detected during more than 25% of the scouting gradient, gradient elution will likely be the best choice for that separation problem. If peaks appear for less than 25% of the scouting gradient, isocratic elution should be preferred, as the selectivity will always be superior under isocratic conditions.
If the sample consists of two distinct groups of compounds, step elution can lead to a good separation results. In this case one starts with isocratic mobile phase elution that renders satisfying separation results for the less retained group of compounds, followed by increasing the elution strength of the mobile phase in one step to such a degree that will separate the more retained group of compounds in an appropriate way. Step gradients can also be used for washing the column after every injection. In this case the elution strength is increased drastically after the last peak has eluted. Strongly retained impurities can then be removed from the column.
| Salt | pKA | Buffer pH range |
equilibrium | UV cut-off |
|---|---|---|---|---|
| Ammonium acetate | 4.76
9.2 |
3.8-5.8
8.2-10.2 |
HAc ↔ Ac−
NH4+ ↔ NH3 |
205 |
| Ammonium formate | 3.8
9.2 |
2.8-4.8
8.2-10.2 |
HCOOH ↔ HCOO−
NH4+ ↔ NH3 |
200 |
| Potassium phosphate | 2.1
7.2 |
1.1-3.1
6.2-8.2 |
H3PO4 ↔ H2PO4−
H2PO4− ↔ HPO42− |
190 |
| Triethylammonium acetate | 4.76
11.01 |
3.8-5.8
10.-12.0 |
HAc ↔ Ac−
Et3NH + ↔ Et3N |
235 |
| Salt | pKA | Buffer pH range |
equilibrium | UV cut-off |
|---|---|---|---|---|
| Acetic acid | 4.76 | - | HAc ↔ Ac− | 205 |
| Formic acid | 3.8 | - | HCOOH ↔ HCOO− | 200 |
| Trifluroacetic acid | 0.3 | - | F3CCOOH ↔ F3CCOO− | 210 |
| Trietylamine | 11.01 | - | Et3NH+ ↔ Et3N | 235 |
| Diethylamine | 10.5 | - | Et2NH2+ ↔ Et2HN | 235 |
| Ammonium hydroxide | 9.2 | - | NH4+ ↔ NH3 | 190 |
It is nearly impossible to foresee which chiral stationary phase will provide the highest selectivity for a certain racemate. Some common racemates that can be separated by our chiral column can be found in the application guide. If the structure of your compound is similar to the compounds shown in the application guide, then there is a good chance that the same column / mobile phase will also provide enantiomeric separation for your compound.
Kromasil AmyCoat and CelluCoat are stationary phases that are very general, thus provide separation for most racemates. These two columns can be used under normal phase, polar mode and reversed phase conditions. For reversed phase applications, specific RP columns should be used. Kromasil AmyCoat and CelluCoat are based on modified polysaccharides that are adsorbed onto a silica matrix. Attention has to be given to the choice of mobile phase solvents in order to avoid desorption of the polysaccharide polymer.
Best results are often obtained when an aprotic polar modifier is used, e.g. ethers or esters incombination with heptane. For basic racemates, acidic and basic additives often lead to significant improvements of the peak shape. The acidic additive (e.g. formic acid or acetic acid) should be used in excess, e.g. 0.1% acid and 0.05% amine (DEA, or TEA).
For detailed information concerning our chiral columns, please consult our chiral brochures.
In order to get the best possible sensitivity, it is important to work with the correct flow cell / column combination. Standard HPLC systems are optimized for 4.0 to 4.6 mm ID columns. If smaller ID columns are used, the standard detector flow cell will likely contribute to the extra column volume to such an extent that peak efficiency is compromised. The table below indicates combinations of detector flow cells and column dimensions:
| Column ID [mm] | Typical flow rate [mL/min] | Flow cell volume [μL] |
|---|---|---|
| 4.6 | 1.0 | 13-15 |
| 4.0 | 0.75 | 13-15 |
| 3.0 | 0.5 | 5-9 |
| 2.1 | 0.2 | 5-9 |
| 1.0 | 0.05 | 1-2 |
| USP | Description | Kromasil products |
| L1 | Octadecyl silane chemically bonded to porous silica | Kromasil 100 Å C18
Kromasil 300 Å C18 Kromasil Eternity C18 Kromasil EternityXT C18 |
| L3 | Porous silica microparticles | Kromasil 60 Å SIL
Kromasil 100 Å SIL Kromasil 300 Å SIL |
| L7 | Octyl silane chemically bonded to porous silica | Kromasil 100 Å C8
Kromasil 300 Å C8 |
| L8 | An essencially monomolecular layer of aminopropylsilane chemically bonded to totally porous silica | Kromasil 100 Å NH2 |
| L10 | Nitrile groups chemically bonded to porous silica microparticles | Kromasil 60 Å CN |
| L11 | Phenyl groups chemically bonded to porous silica particles | Kromasil 100 Å Phenyl
Kromasil Eternity PhenylHexyl Kromasil EternityXT PhenylHexyl |
| L13 | Trimethylsilane chemically bonded to porous silica microparticles | Kromasil 100 Å C1 |
| L26 | Butyl silane chemically bonded to totally porous silica particles | Kromasil 100 Å C4
Kromasil 300 Å C4 |
Certain buffers or organic solvents interfere with UV detection. Only mobile phases with a UV cut-off below the detection wavelength will not compromise the signal sensitivity.
The tables below show the UV cut-off of the most commonly used buffers, solvents and additives in HPLC.
| pH value | Buffer | UV cut-off [nm] |
|---|---|---|
| 2.0 – 3.0 | Phosphate | 210 |
| 3.5 – 5.5 | Acetate | 240 |
| 4.0 – 6.0 | Citrate | 250 |
| 6.0 – 8.5 | Phosphate | 210 |
| 7.0 – 9.0 | TRIS | 225 |
| 8.0 – 10.5 | Borate | 210 |
| Solvent | UV cut-off [nm] |
|---|---|
| Acetone | 330 |
| Acetonitrile | 190 |
| Dichloromethane | 233 |
| Dimethoxy sulfoxide | 268 |
| Dioxan | 215 |
| Ethanol | 210 |
| Ethylacetate | 256 |
| Hexane | 195 |
| Heptane | 200 |
| Methanol | 205 |
| Methyl-ethyl ketone | 329 |
| Methyl t-butyl ether | 210 |
| N,N-Dimethylformamide | 268 |
| Propan-2-ol (IPA) | 205 |
| Tetrahydrofuran | 210 |
| Toluene | 284 |
| Additive | UV cut-off [nm] |
|---|---|
| Acetic acid | 230 |
| Diethylamine | 210 |
| Formic acid | 210 |
| Triethylamine | 235 |
| Trifluoroacetic acid | 210 |
Peak tailing can occur due to numerous reasons. The problem can be identified according to the following scheme:
-
Mass overload: when injecting less sample amount (mass) either the peak becomes
more symmetrical or resolves into two separate peaks.
Corrective action: use a more dilute injection sample.
-
Secondary interactions: when injecting a neutral compound (acetophenone, toluene)
the peak becomes symmetrical
Corrective action: adjust the mobile phase pH in order to render neutral analytes.
For larger ID columns (>10 mm) radial temperature gradient can also cause peak tailing. In order to avoid such, it is recommended to use a column oven. The column should be 1-2°C warmer than the mobile phase in order to compensate for the friction heat generated in the core of the column.
If neither of the above applies, the tailing can also be caused by irregularities in the column packing process or by a partially clogged inlet frit leading to an inhomogeneous flow profile.
1. Injection amount / injection volume
Overloading by mass or volume as well as the wrong sample solvent can decrease the efficiency of the column significantly and therewith impair the results of the separation. The injection volume should not exceed 10% of the flow rate (e.g. 1 ml/min ? injection volume < 100 µL. Mass overloading is obtained as soon as the linear part of the adsorption isotherm is surpassed. This border is individual, but as a rule of thumb, no more than 0.01 mg sample should be loaded per ml column volume (e.g. max 22 µg on 4.6 × 250 mm column). The sample mass may however be increased if necessary for detection reasons. In the sample solvent, the amount of the strong mobile phase component (alcohol, acetonitrile in RP, or alcohol, ethyl acetate etc. in NP), should not exceed the amount in the mobile phase.
2. Extra column volume / dead volumes
Poor plate counts can often be attributed to excessive dead volume. The dead volume is defined as the sum of the volume contributed by the sample injector, including the sample loop, the tubing connecting the sample injector to the column, the tubing connecting the column to the detector flow cell, the detector flow cell, plus any volume added by fittings, connectors, and in-line filters. Tubing with 0.010" ID is commonly found in HPLC systems. If you are using narrow bore (< 3.0 mm ID) columns, or, you can enhance your system´s performance by replacing the 0.010" ID tubing with 0.007" ID tubing to reduce system dead volume. Tubing with 0.005" ID can further reduce dead volume, but at a cost of inconvenience. Tubing with IDs less than 0.007" restricts flow rate and may have problems with buffer salt precipitation. If you use a variety of brands of HPLC columns, make sure that the fittings used to connect to the column are properly matched. If your system has stainless steel tubing and fittings, select the proper fittings for each brand of column that you use and install those fittings prior to connecting the column. If your system has PEEK tubing and fittings, it is a good idea to install a new PEEK fitting on a freshly cut piece of PEEK tubing with each column change to insure a proper fit.
Split Peaks can be caused by:
- Column contamination
- Partially plugged frit
- Column Void
- Injection Solvent Effects
- Co-eluting compounds
Column contamination or partially clogged frits are generally caused when unfiltered samples are injected or when the sample solvent is different to the mobile phase. If the latter is the case, constituents of the sample may precipitate at the column inlet upon mixing with the mobile phase or by contact with the metallic surface of the frit.
A column void is formed either by hydrodynamic stress (high flow rate, high viscosity of the mobile phase) of a poorly packed column, by mechanical impact (the column was dropped on the floor) or by chemical dissolution of the packing material, e.g. pH > 12.
When using injection solvents that have higher elution strength than the mobile phase split or broader peaks can appear. The effect is most pronounced for early eluting peaks.
Split peaks can also occur by either two compounds that are almost co-eluting. If this is the case, the peaks should separate better if a column with higher efficiency (theoretical plates) is used. This can either be achieved by smaller particle size or a longer column. Also higher temperature and/or a less viscous mobile phase may promote a better separation.
Moreover, one compound can elute in two states, e.g. if not sufficient ion-pairing reagent is present in the mobile phase or the buffer capacity is deficient.
The reason for the variability in retention time in normal phase HPLC is the strong dependence of the retention on the content of highly polar mobile phase constituents, especially water. Even if no water is added to the mobile phase on purpose, small quantities will always be dissolved even in very apolar solvents. Furthermore, bare silica is extremely hygroscopic, thus water will be adsorbed on the surface. Hence, the water content in a normal phase system can vary significantly and is seldom controlled. Best results, e. g. reproducible retention times, are obtained when working with half-saturated mobile phases. A straight forward way to obtain a half saturated mobile phase is to divide the mobile phase in to equal parts. To one part (e.g. 500 mL), one adds 2-3 mL water and allows the mixture to stir for about 30 min. Thereafter the excess water is removed (separating funnel), and the dry part (500 mL) is added to the saturated portion.
Even if a half saturated mobile phase will reduce the time to reach equilibrium in the column to a large extent. The equilibration of a normal phase column, especially when dealing with bare silica, can still take hours. Furthermore, polar modifier stationary phases, such as cyano or diol, are generally much less prone to variations in the water content of the mobile phase and can be equilibrated faster.
Some examples of solubility data of water in organic solvents at 25°C.:
- Heptane: 0.0091% (w/w)
- Ethyl acetate: 2.94% (w/w)
- Toluene: 0.0334% (w/w)
- MTBE: 1.50% (w/w)